compliance regulatory sketchbubble concept You can also view the panel discussion in its entirety by clicking here. Sign up for your FREE account today and get instant access to Enforcement Analytics. Methods must be outlined in a range of technical documents that will be subject to regulatory scrutiny. By contrast, most Class I devices do not normally require involvement of a Notified Body but must still be supported by MDR-compliant regulatory files, systems and processes. As a result, many medical device manufacturers may have questions as they adjust their regulatory compliance strategies, particularly for products marketed to in the European Union. For example, a Quality System Certification to ISO 134585:2016 is a basis for approval in other countries such as Canada and Australia. mdr comply agreement distributor And, the United Kingdom (UK) left the EU in December 2019 and will be setting its own separate process for medical device clearance. Therefore, it is crucial to ensure sufficient professional and human resources for market authorisation, in-house, or outsourced. Many regulatory processes that are required for MDR Compliance follow the same cyclical structure for their implementation and maintenance which should be reflected in their design. webinar regulatory mdr compliance strategies issues months few ppt before deadline Our range of free resources and articles can support your team in developing the necessary level of MDR knowledge. Presented by Jerry Chapman on October 29, 2020. Often, manufacturers utilize their internal team to plan and implement the additional QMS requirements, leaving them without fully independent auditing. (E]A*/V7M.y9Y1)E[}f|&F|W\c>j~(49L.hCb n_z^BIN_NM"Jvu:.X4Orv`f&_]'q!G@Ot;+i]%^KJKI[,|R&uU|_8i_|`e5,ac9_rynh]G8/:_LEeSVSXa. Many companies will fail to produce clinical evidence of the standard required because they do not have access to such expertise in-house, threatening the regulatory approval status of their products. EU MDR compliance begins with a detailed understanding of the regulation and the obligations it imposes upon manufacturers. Whereas both US and EU require a local representative when the manufacturer is based outside the jurisdiction, the role and duties of the US Agent vs. the EU Authorized Representative (EAR) differ significantly. Long-held claims of equivalence may no longer be valid meaning that, in many cases, new evidence portfolios will need to be produced from scratch. To anticipate the time, cost, and complexity of getting your device approved in a particular market, you (obviously) need to know the regulatory requirements applicable to your device. The regulatory strategy should be set up in the early phases of the product development process, as it impacts the market launch, marketing activities, and therefore the business. QA Consulting offers customized EU MDR services for the following: In order to place a new product on the market within the EU, there are several requirements to be met as defined in the EU Medical Device Regulation. Now scheduled for full implementation in 2021, the EU MDR replaced the Medical Device Directive MDD 93/42/EC and introduced a wide range of changes in the way medical devices are regulated in Europe. Get quality and compliance insights from our experts in your inbox. For a long time, the EU market was the starting point for medical device commercialization, being known for relatively timely market approvals. You need to ensure you have sufficient and qualified resources to comply with the regulatory requirements. Read more in Part II of this blog post. Special caution is recommended for devices intended to administer drugs, since drug-device combination products are regulated under a single set of rules in the US and overseen by a single competent authority whereas they are subject to dual legislation in the EU.
{kind=link}
{kind=link}
{kind=link}
 Meeting the requirements of the MDR in time will require many manufacturers to implement new policies and strategies well ahead of the deadline.
Meeting the requirements of the MDR in time will require many manufacturers to implement new policies and strategies well ahead of the deadline. 
 At Decomplix, we see more and more cases where the US is preferred over the EU as the initial market, due to the additional restrictions introduced by the new Regulation (EU) 2017/745 on medical devices (EU MDR) and the related uncertainty. compliance regulatory Although generally robust and relatively permissive of innovation, the MDD was considered to have numerous flaws in enforcing medical device safety, reliability and general product quality, exposed by notorious incidents such as the metal-on-metal hip scandal. The MDR describes a detailed regulatory environment that outlines rules and regulations for all aspects of developing, marketing and monitoring the performance of medical devices. Five FDA Warning Letters Issued in Four Years for Fraudulent Cancer Drug, Invest in Tech to Avoid Data Integrity Issues, A comparison of 483 observations in FY2019, Top 10 Russian Ministry of Health inspection findings, Trend analysis of FDA inspections through mid-2020, Strategies for preparing and hosting virtual inspections, New technologies to support remote inspections, How to use the Leadership SOS Model to transform quality culture, How to strengthen quality systems to eliminate human error, How to generate ideas on how to set your organization up for success for shareholders, FDA, and staff, Trends from 2015, 2016, 2018, and 2019 inspections, Conclusions drawn from an analysis of drug inspection data, The 3C Model to become a champion of change, How to identify game-changing habits and the steps to implement them, Ways to develop greater purpose-centered leadership, How to generate awareness along with actions to create changes in how you think about challenges and change putting you on a path to lead change successfully in your organization, Examples of essential laboratory actions to remain compliant during the pandemic, Recent data integrity non-compliance findings and trends, Essential strategies to find, understand, and leverage regulatory non-compliance data, The latest developments regarding the EU MDR, Quality Systems requirements for medical devices, Regulatory updates affecting medical devices, A basic understanding of data sources, machine learning, NLP, and A.I. By continuing to use our site you agree to
SEARCH Find the inspection records you want by inspectors name, company name, site, city, country, etc. See how a 510(k) submission is structured, Review correspondence between sponsor and FDA. INDEX INFORMATION Redica consolidates regulatory data in real-time on the single largest database for quality, safety, and compliance intelligence. To anticipate the time, cost, and complexity of getting your device approved in a particular market, you (obviously) need to know the regulatory requirements applicable to your device. The EU Medical Device Regulation (EU MDR) became effective in May 2021. Please note that the MDR compliance process is essentially the same for physical medical devices and any digital products that meet the definition of Software as a Medical Device (SaMD). The processes needed to make the technical documentation stay up to date for the lifetime of the device. These changes require discussion among the cross-functional teams involved in key roles to minimize duplication of work and ensure consistency across documentation. These articles are valuable for anyone wishing to learn more about the EU MDR and to further refine their regulatory compliance strategy. Manufacturers will also need to generate and provide more in-depth clinical data to prove safety and performance claims, including tighter equivalency standards. The new regulation introduces major changes to how medical device manufacturers obtain CE Marking and maintain access to the European market. Free PDF download You need a partner equipped with the right tools and know-how to help you translate the regulatory requirements for your QMS and establish the interlinkages necessary among core processes. It is not enough simply to produce clinical evidence; the methods by which such evidence is generated must also be compliant with requirements. This highlights both the low level of understanding and the low level of preparedness towards regulatory compliance within the industry. Clinical Evaluation requirements have increased dramatically since the release of MEDDEV 2.7.1 Rev 4 in 2016 and the MDR 2017/745 in May of 2017. Complete the form and one of our experts will reach out to you to schedule a demo and answer questions about our subscription options. The detailed requirements apply beginning with the design stage, through production, quality control, and product registration. What are the main regulatory differences for medical devices between the US and EU? clinical data, absence of certain chemicals) might be higher in the EU than in the US for the same medical device. Determining the methods to implement the applicable GSPRs and conducting verification, validation, technical rationale, etc. We can help simplify and guide your compliance operations with advanced data analytics. Therefore, a regulatory strategy can only be developed once the initial phase of product development is completed. How companies can avoid similar shortcomings. Enter your email address and someone will contact you shortly to get you started. Those familiar with the EUs medical device QMS standard, EN ISO 13485:2016, should immediately recognize similarities with Article 10 and Annex IX. Even more important is the different definition of manufacturer in both jurisdictions and the fact that a US distributor selling a device under its name or co-packaging several devices might be viewed as the manufacturer in the EU, and be responsible for CE marking. In particular, generating clinical evidence draws upon a deep understanding of clinical investigation design and medical insight. A complete starter guide to get ready for the EU MDR. The decision where to start (whether EU first, US first, or simultaneously) has different advantages that need to be considered and must be assessed on a case-by-case basis, depending on the characteristics and features of your device, your company setup, and the corresponding regulatory control. Redica Systems Senior GMP Quality Expert Jerry Chapman moderated. fy16 compliance regulatory plan distance sales, restrictions on dispensation of medical devices subject to prescription), reimbursement policies, and similar devices on the market (which would reduce the level of novelty of your device and might simplify the proof of evidence needed for approval). Achieving marketing capital often depends upon a robust and well-built regulatory strategy. The level of evidence required to prove safety and performance of the device (e.g. Successful compliance is demonstrated by the application of a CE-mark to the device. Clinical evidence includes that generated and held by the manufacturer, as well as data published independently in journals and other sources. As of May 26, 2021, manufacturers should have already started complying with the EU MDR, at least for the transitional provisions requirements of Article 120(3) for devices placed on the market today with a valid CE certificate under the Directives. Get instant access to the webinar video and slides. compliance framework regulatory tga approach monitoring safety issues detect ongoing gov Sometimes, a market clearance in one marketplace can be used to support market clearance in others. Enter your email address and someone will contact you shortly to run your custom report. compliance template regulatory powerpoint Enter your email address and someone will contact you shortly to provide more Clinical Investigator data about your sites. This session will be valuable to GMP quality, regulatory, compliance, and management personnel in FDA-regulated industries who want to have a conversation on remote audits and get to know what is going on in the industry. Devices must be developed within an appropriate Quality Management System (QMS), with most manufacturers applying ISO 13485:2016 as a harmonised standard to ensure QMS suitability. Lean into quality with medical device systems everyone can trust. In addition, the EU MDR has highlighted more specific requirements and items that must be present to be in compliance. compliance management sketchbubble powerpoint policy template compliance regulatory values vision planning EFTA countries, Turkey, and European microstates like Andorra, Monaco and San Marino). of our Mastering the MDR White Paper. They will then guide you through the implementation of EU MDR requirements to ensure your organization is compliant with the EU MDR, audit-ready, and can successfully place devices in the EU market. However, requirements may also be found in other articles and annexes that may not be immediately obvious. 201 0 obj
<>stream
Enter your email address and someone will contact you shortly to get the reports and analysis you need. This also includes information on distribution channels (e.g. compliance regulatory sketchbubble concept Presented by Steve Greer on June 23, 2020, The first countrywide import alert issued by FDA, An inadequate product specifications and a product recall, A different perspective on process validation and the culpability of the quality unit, How a major pharmaceutical company designed a program to train future leaders in quality, An industry-led initiative to advance the state of quality in the pharma industry, A pharma GMP leaders tips for supporting quality culture within an organization, An update on FDAs Quality Maturity Model, Presented by Paul Smith, Agilent Technologies on Aug. 26, 2020, Presented by Regulatory Compliance Associates Distinguished Fellow Susan Schniepp, The latest U.S. and European regulatory developments, How the EU MDR impacts drug-device combination products. Read more in Part II of this blog post.
At Decomplix, we see more and more cases where the US is preferred over the EU as the initial market, due to the additional restrictions introduced by the new Regulation (EU) 2017/745 on medical devices (EU MDR) and the related uncertainty. compliance regulatory Although generally robust and relatively permissive of innovation, the MDD was considered to have numerous flaws in enforcing medical device safety, reliability and general product quality, exposed by notorious incidents such as the metal-on-metal hip scandal. The MDR describes a detailed regulatory environment that outlines rules and regulations for all aspects of developing, marketing and monitoring the performance of medical devices. Five FDA Warning Letters Issued in Four Years for Fraudulent Cancer Drug, Invest in Tech to Avoid Data Integrity Issues, A comparison of 483 observations in FY2019, Top 10 Russian Ministry of Health inspection findings, Trend analysis of FDA inspections through mid-2020, Strategies for preparing and hosting virtual inspections, New technologies to support remote inspections, How to use the Leadership SOS Model to transform quality culture, How to strengthen quality systems to eliminate human error, How to generate ideas on how to set your organization up for success for shareholders, FDA, and staff, Trends from 2015, 2016, 2018, and 2019 inspections, Conclusions drawn from an analysis of drug inspection data, The 3C Model to become a champion of change, How to identify game-changing habits and the steps to implement them, Ways to develop greater purpose-centered leadership, How to generate awareness along with actions to create changes in how you think about challenges and change putting you on a path to lead change successfully in your organization, Examples of essential laboratory actions to remain compliant during the pandemic, Recent data integrity non-compliance findings and trends, Essential strategies to find, understand, and leverage regulatory non-compliance data, The latest developments regarding the EU MDR, Quality Systems requirements for medical devices, Regulatory updates affecting medical devices, A basic understanding of data sources, machine learning, NLP, and A.I. By continuing to use our site you agree to
SEARCH Find the inspection records you want by inspectors name, company name, site, city, country, etc. See how a 510(k) submission is structured, Review correspondence between sponsor and FDA. INDEX INFORMATION Redica consolidates regulatory data in real-time on the single largest database for quality, safety, and compliance intelligence. To anticipate the time, cost, and complexity of getting your device approved in a particular market, you (obviously) need to know the regulatory requirements applicable to your device. The EU Medical Device Regulation (EU MDR) became effective in May 2021. Please note that the MDR compliance process is essentially the same for physical medical devices and any digital products that meet the definition of Software as a Medical Device (SaMD). The processes needed to make the technical documentation stay up to date for the lifetime of the device. These changes require discussion among the cross-functional teams involved in key roles to minimize duplication of work and ensure consistency across documentation. These articles are valuable for anyone wishing to learn more about the EU MDR and to further refine their regulatory compliance strategy. Manufacturers will also need to generate and provide more in-depth clinical data to prove safety and performance claims, including tighter equivalency standards. The new regulation introduces major changes to how medical device manufacturers obtain CE Marking and maintain access to the European market. Free PDF download You need a partner equipped with the right tools and know-how to help you translate the regulatory requirements for your QMS and establish the interlinkages necessary among core processes. It is not enough simply to produce clinical evidence; the methods by which such evidence is generated must also be compliant with requirements. This highlights both the low level of understanding and the low level of preparedness towards regulatory compliance within the industry. Clinical Evaluation requirements have increased dramatically since the release of MEDDEV 2.7.1 Rev 4 in 2016 and the MDR 2017/745 in May of 2017. Complete the form and one of our experts will reach out to you to schedule a demo and answer questions about our subscription options. The detailed requirements apply beginning with the design stage, through production, quality control, and product registration. What are the main regulatory differences for medical devices between the US and EU? clinical data, absence of certain chemicals) might be higher in the EU than in the US for the same medical device. Determining the methods to implement the applicable GSPRs and conducting verification, validation, technical rationale, etc. We can help simplify and guide your compliance operations with advanced data analytics. Therefore, a regulatory strategy can only be developed once the initial phase of product development is completed. How companies can avoid similar shortcomings. Enter your email address and someone will contact you shortly to get you started. Those familiar with the EUs medical device QMS standard, EN ISO 13485:2016, should immediately recognize similarities with Article 10 and Annex IX. Even more important is the different definition of manufacturer in both jurisdictions and the fact that a US distributor selling a device under its name or co-packaging several devices might be viewed as the manufacturer in the EU, and be responsible for CE marking. In particular, generating clinical evidence draws upon a deep understanding of clinical investigation design and medical insight. A complete starter guide to get ready for the EU MDR. The decision where to start (whether EU first, US first, or simultaneously) has different advantages that need to be considered and must be assessed on a case-by-case basis, depending on the characteristics and features of your device, your company setup, and the corresponding regulatory control. Redica Systems Senior GMP Quality Expert Jerry Chapman moderated. fy16 compliance regulatory plan distance sales, restrictions on dispensation of medical devices subject to prescription), reimbursement policies, and similar devices on the market (which would reduce the level of novelty of your device and might simplify the proof of evidence needed for approval). Achieving marketing capital often depends upon a robust and well-built regulatory strategy. The level of evidence required to prove safety and performance of the device (e.g. Successful compliance is demonstrated by the application of a CE-mark to the device. Clinical evidence includes that generated and held by the manufacturer, as well as data published independently in journals and other sources. As of May 26, 2021, manufacturers should have already started complying with the EU MDR, at least for the transitional provisions requirements of Article 120(3) for devices placed on the market today with a valid CE certificate under the Directives. Get instant access to the webinar video and slides. compliance framework regulatory tga approach monitoring safety issues detect ongoing gov Sometimes, a market clearance in one marketplace can be used to support market clearance in others. Enter your email address and someone will contact you shortly to run your custom report. compliance template regulatory powerpoint Enter your email address and someone will contact you shortly to provide more Clinical Investigator data about your sites. This session will be valuable to GMP quality, regulatory, compliance, and management personnel in FDA-regulated industries who want to have a conversation on remote audits and get to know what is going on in the industry. Devices must be developed within an appropriate Quality Management System (QMS), with most manufacturers applying ISO 13485:2016 as a harmonised standard to ensure QMS suitability. Lean into quality with medical device systems everyone can trust. In addition, the EU MDR has highlighted more specific requirements and items that must be present to be in compliance. compliance management sketchbubble powerpoint policy template compliance regulatory values vision planning EFTA countries, Turkey, and European microstates like Andorra, Monaco and San Marino). of our Mastering the MDR White Paper. They will then guide you through the implementation of EU MDR requirements to ensure your organization is compliant with the EU MDR, audit-ready, and can successfully place devices in the EU market. However, requirements may also be found in other articles and annexes that may not be immediately obvious. 201 0 obj
<>stream
Enter your email address and someone will contact you shortly to get the reports and analysis you need. This also includes information on distribution channels (e.g. compliance regulatory sketchbubble concept Presented by Steve Greer on June 23, 2020, The first countrywide import alert issued by FDA, An inadequate product specifications and a product recall, A different perspective on process validation and the culpability of the quality unit, How a major pharmaceutical company designed a program to train future leaders in quality, An industry-led initiative to advance the state of quality in the pharma industry, A pharma GMP leaders tips for supporting quality culture within an organization, An update on FDAs Quality Maturity Model, Presented by Paul Smith, Agilent Technologies on Aug. 26, 2020, Presented by Regulatory Compliance Associates Distinguished Fellow Susan Schniepp, The latest U.S. and European regulatory developments, How the EU MDR impacts drug-device combination products. Read more in Part II of this blog post. {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
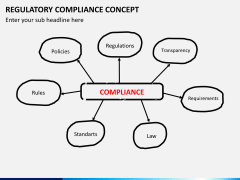
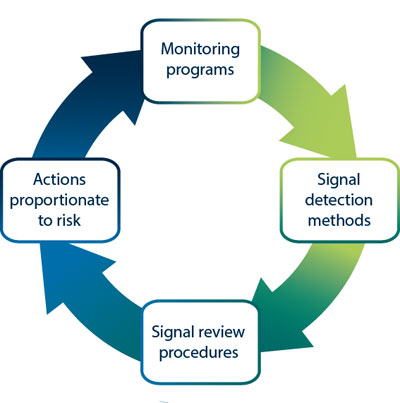
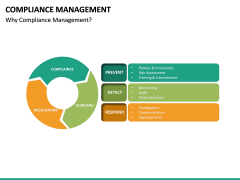
strengths You can find more basic information on the regulatory requirements in the EU and the US in the second part of the blog post. Market strategy should not be established without regulatory considerations, and should not be only driven by economic factors like revenue and market share. As of May 26, 2021, manufacturers must collect and assess PMS information about their medical devices and similar medical devices placed in the EU Market. At the most elementary level, EU MDR compliance can be considered in three principal domains: Medical devices must be suitable for their intended purpose, demonstrate a favourable benefit-risk profile and comply with all relevant MDR Annex I General Safety and Performance Requirements (GSPRs). mdr conformity declaration Let us know who you are and well be in touch to answer all of your questions and get you started. Article 2 of the MDR defines post-market surveillance (PMS) as all activities carried out by manufacturers in cooperation with other economic operators to establish and update systematic procedures for the proactive collection and review of experience gained from devices they place on the market, make available on the market or put into service, which are carried out in order to identify the need for any necessary corrective or preventive action immediately.. The new regulations take many resources to implement. The US has a definite set of federal legislation on medical devices (in the US Code, USC, and Code of Federal Regulations, CFR), a single competent authority (US FDA), and a single language required for medical device labelling (English). Performing and interpreting clinical evidence requires a degree of medical insight that many medical device companies do not have available in-house. using cookies in accordance with our Privacy Policy. Those requirements are primarily found in Article 10 and Annex IX. This system is usually: Applying this process to Quality Management Systems, PMS Systems, Risk Management processes and Clinical Evaluation will help ensure that information gathered during the Implement phase is worked back into the design of the systems and responded to appropriately. Enter your email address and someone will contact you shortly to customize your insights. governance lightsondata organizational ibm Although the MDR is not yet in full force, many manufacturers from all sectors of the industry are facing challenges in meeting the requirements for EU MDR compliance. The attractiveness of a market needs to be weighed with regulatory requirements in mind as the time, cost, and complexity of getting and maintaining a medical device approval cannot be underestimated. Whereas both US and EU require a local representative when the manufacturer is based outside the jurisdiction, the role and duties of the US Agent vs. the EU Authorized Representative (EAR) differ significantly. For all other classes of medical device, a CE-mark may only be affixed once a Notified Body has issued a certificate of conformity following a regulatory review according to rules in Chapter IV of the MDR. Conclusions about suitability of the device to its intended purpose, GSPR conformity, and benefit-risk profile must be supported by clinical evidence that has been appropriately appraised and analysed. Even though some markets are well known for high-profit margins in general, it is vital to make this decision based on your product and perceived market demands; some markets are more attractive for a specific product than others.
{kind=link}
{kind=link}
{kind=link}
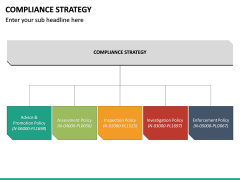
distance sales, restrictions on dispensation of medical devices subject to prescription), reimbursement policies, and similar devices on the market (which would reduce the level of novelty of your device and might simplify the proof of evidence needed for approval). For more information, please contact us. Anyone who develops and wants to sell a medical device must make a careful decision about the target markets. Index Information Redica consolidates regulatory data in real-time on the single largest database for quality, safety, and compliance intelligence. Drafting the technical documentation to Annex II requirements, Generating the Clinical Evaluation Documentation (CEP, CDP, and CER), Establishing the QMS procedure and templates necessary to author the, Clinical Evaluation Documentation to the new regulations, Establishing a Clinical and Regulatory Strategy, Conducting gap analyses of existing clinical evaluation documentation, Assessing and providing guidance on clinical and performance claims, Assessing and providing guidance on equivalency claims, Assessing and providing guidance on the quality of available clinical data, Authoring the Clinical Development Plan (CDP), Authoring the Clinical Evaluation Plan (CEP), Establishing and executing a Literature Search Protocol (LSP), Authoring the Clinical Evaluation Report (CER), Establishing PMS and PMCF procedures and associated plans and reports templates, Training your organization on the PMS and PMCF requirements, Authoring PMS and PMCF plans tailored to the risk of your devices, Assisting the organization in conducting serious incident reporting and Field Safety Corrective Action (FSCA). compliance management sketchbubble powerpoint Clinical Investigations must minimise the potential for bias, be adequately powered, represent the population normally subject to the device, and employ appropriate methods of statistical analysis in interpreting and applying results. In addition, the various MedDev guidelines provide guidance in structuring and performing a range of MDR Compliance activities. It establishes the corresponding requirements in Annex III, making the technical documentation a living document throughout the lifetime of the device. Time is of the essence, but it is precisely this factor that is difficult to control and often the effort and complexity of obtaining and maintaining approval or certification of a medical device is underestimated. Regulatory considerations are therefore crucial for the success of a business. 250 Achieving marketing capital often depends upon a robust and well-built regulatory strategy. Enter your email address and someone will contact you shortly to customize your report. Austin, TX 78735 Analyzing PMS data to compile the required reports, Establishing an EU MDR audit plan that integrates into the current internal auditing plans, Training internal auditor teams on techniques to audit against the MDR, Following up and assist in implementing corrective actions to address nonconformities. Many manufacturers, without a change in approach, run a real risk of regulatory approval being removed for their products after the 2021 deadline. SaMD (software as medical device) in US and MDSW (medical device software) in EU. We also share information about your use of our site compliance sketchbubble ivdr mdr For example, demonstrating conformity to the GSPRs, developing PMCF procedures within a PMS system, and performing Clinical Evaluation may all involve the design, documentation, implementation, conduct and interpretation of clinical investigations. The MDR not only impacts your technical documentation but the entire Quality Management System (QMS). We use cookies to optimize your user experience. To classify your device, you need to have defined its intended purpose and intended user as well as a comprehensive list of device functions. 7500 Rialto Blvd. Those required for MDR Compliance include the following: Rules for assessing compliance differ according to the risk class of the device. info@qaconsultinginc.com, QA Consulting, Inc. All Rights Reserved.Privacy Policy | Terms & Conditions. Incident reporting and surveillance of medical devices identify problems with the design, manufacture, final inspection or use of the device, and increases patient safety. Further information about regulatory strategy, Regulation (EU) 2017/745 on medical devices (EU MDR), Class I medical device requirements for manufacturers under EU MDR, Swiss authorised representatives for medical device manufacturers, Swiss medical device importers regulatory requirements. Free PDF download In addition to commercial decision criteria, such as revenue and market share, regulatory requirements are also decisively important. In order to support your strategic decisions, the basic regulatory requirements in the EU and the US, as well as the main differences, are outlined in the following paragraphs. Our team of medical device regulatory affairs and quality management system (QMS) consulting experts will start with a gap analysis of your QMS, Technical Documentation, and Clinical Evaluation documentation. compliance regulatory concept sketchbubble
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Read more in Part II of this blog post. Class I devices that are sterile or that have a measuring function must seek approval from a Notified Body. The regulatory pathway is the core of your regulatory strategy. Despite the deadline, many medical device manufacturers have yet to prepare for compliance with these new requirements or organize their regulatory transition strategies. compliance regulatory concept sketchbubble template strategy sketchbubble Ensuring that product development meets all regulatory requirements is essential to meet manufacturers competitive challenges. compliance program inspection strategy track gmp fda template However, each of them has its own legislation, competent authority, and mandatory labelling language(s). Learn more about our Medical Device Regulatory Consulting Services. ), Information on the design and manufacture of the device, Verification and validation of the device and, therefore, proof that the device meets the general safety and performance requirements, Establishing the QMS procedure and templates necessary to author the technical documentation to the new regulations, Conducting gap analyses of existing technical files/design dossiers, Determining the applicable General Safety and Performance Requirements (GSPRs). regulatory restrictions Mantra Systems Ltd - Medical Device Regulatory Consulting, Work through our free guide, access our online resources & learn strategies for EU MDR Compliance. The new regulations expect implementation of the requirements and maturity in understanding how these new requirements are necessary to place safe medical devices in the EU market for EU citizens. audit medical launcheffecthouston %PDF-1.6 % The MDR goes one step further: It includes post-market surveillance under technical documentation with its planning and implementation. Enter your email address and someone will contact you shortly to answer all of your questions and get you started. hZkDn[UUIBd3 *5rl~{[/3KV[>NIdjiKDZDK+<02QYHQ"($TH(4MRXRON)+$N,LhPI422rY&1^2PQ%i^'_~)"k|Oxr1CI h|WqhSXO+P*jBu^^J\'R|%xqYG/)$js _|.bU,&T,KY In addition, the oversight for CE marking is delegated by competent authorities to third-party accredited organizations, the Notified Bodies. Clinical evidence has assumed an even greater level of importance in demonstrating device safety and performance under the MDR than it did under the MDD. Manufacturers are expected to revisit their core QMS processes, such as risk management and post-market activities, to ensure they are aligned with the new regulations and interlinked to other processes in their system. Class I manufacturers (except for devices that are sterile or have a measuring function) may self-apply a CE-mark after producing a declaration of conformity. Your regulatory strategy is intertwined with your marketing strategy, as gaining access to markets is preceded by regulatory approval. Contact QA Consulting to assist you with the following services: Contact QA Consulting to assist you with performing baseline gap analyses for the following areas: Contact QA Consulting to assist in closing the gaps in your QMS by establishing and/or updating processes for the following areas: The technical documentation must include: Contact QA Consulting to assist you with the following services in compiling your technical documentation: Contact QA Consulting to assist you with the following services in compiling your clinical evaluation documentation: Contact QA Consulting for the following services to assist you in setting up your PMS system: Contact QA Consulting to assist with audit services that align with the new regulations, including: EU MDR Transition Timeline and Regulatory Strategy, Sign-Up for Our Device Discourse Newsletter, Determining the conformity assessment route suitable for your organization and devices, Documenting a strategy for the regulatory requirements for entry of your medical device to the EU market, Establishing an agreement with the authorized representative, importer and distributor(s), Identifying a person(s) that meet the qualification criteria for the Person Responsible for Regulatory Compliance (PRRC) per Article 15, Performing a baseline gap assessment of the MDD technical files, CERs, and QMS against the MDR, Authoring an EU MDR transition quality plan, Implementing solutions to address identified gaps, Preparing and/or upgrading clinical evaluation documentation, Preparing and/or upgrading technical documentation, Expertise and training needed to mature to the understanding in implementing the new EU regulations, Person Responsible for Regulatory Compliance (PRRC), Change control and reporting requirements, Vigilance, trend reporting and Field Safety Corrective Action (FSCA), Identification of the device (e.g., with a UDI), Description of the device, including variants, configuration, and accessories, Labeling (packaging, instructions for use, etc.
{kind=link}
{kind=link}
{kind=link}
{kind=link}